Akromegali adalah penyakit yang berlaku apabila kelenjar pituitari mengeluarkan terlalu banyak hormon pertumbuhan (growth hormone), biasanya akibat tumor pada kelenjar tersebut. Ia menyebabkan tangan, kaki, dan muka membesar secara perlahan-lahan, serta boleh mendatangkan pelbagai komplikasi serius jika tidak dirawat. Rawatan utama adalah pembedahan, diikuti dengan ubat-ubatan atau radioterapi bergantung kepada keperluan pesakit.
- Akromegali disebabkan oleh adenoma pituitari yang menghasilkan hormon pertumbuhan secara berlebihan.
- Gejala utama termasuk pembesaran tangan, kaki, rahang, dan perubahan pada muka yang berlaku secara beransur-ansur.
- Ia adalah penyakit yang jarang berlaku, dengan anggaran 4–6 kes baru bagi setiap juta penduduk setahun [6].
- Diagnosis dibuat melalui ujian darah (IGF-1 dan ujian penekanan glukosa) serta MRI pituitari.
- Rawatan boleh melibatkan pembedahan, ubat-ubatan, dan radioterapi — pilihan bergantung kepada saiz tumor dan respons rawatan.
- Kawalan penyakit yang ketat penting untuk mengurangkan kadar kematian [5].
Apakah Akromegali?
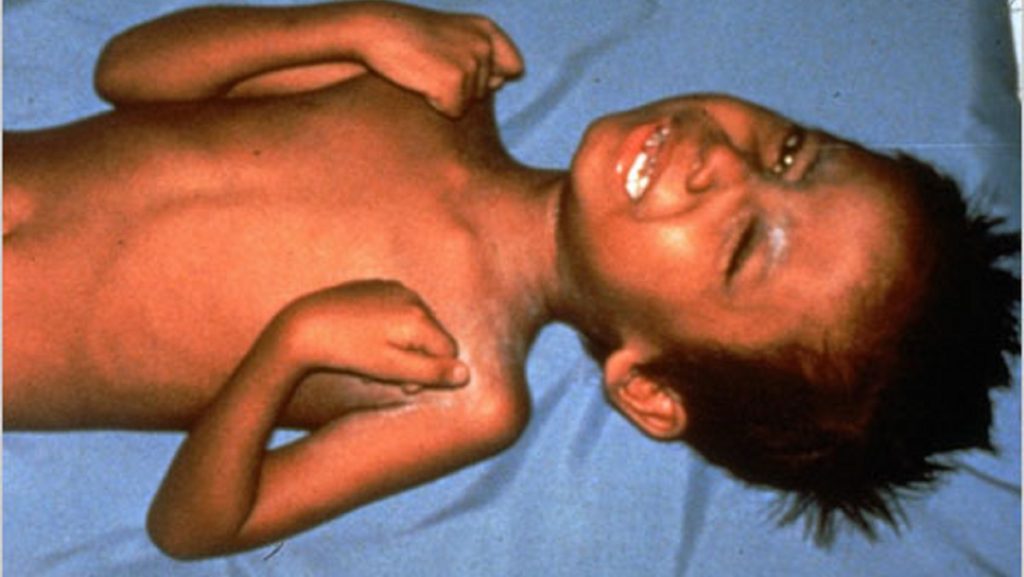
Akromegali adalah penyakit yang berlaku apabila tubuh menghasilkan terlalu banyak hormon pertumbuhan (growth hormone atau GH) selepas seseorang itu sudah matang sepenuhnya. Hormon ini penting semasa zaman kanak-kanak untuk pertumbuhan badan, tetapi apabila ia berlebihan pada orang dewasa, ia menyebabkan tulang dan tisu lembut pada bahagian tertentu badan terus membesar walaupun usia sudah meningkat.
Nama “akromegali” berasal daripada perkataan Greek — akron bermaksud hujung atau anggota badan, dan megas bermaksud besar — menggambarkan ciri paling ketara penyakit ini: pembesaran pada hujung badan seperti tangan dan kaki.
Penyakit ini jarang berlaku, dengan anggaran 4 hingga 6 kes baru bagi setiap juta penduduk setahun [6]. Walaupun jarang, ia membawa komplikasi serius termasuk masalah jantung, kencing manis, dan tekanan darah tinggi jika tidak diurus dengan baik.
Bagaimana Akromegali Berlaku?
Kelenjar pituitari — kelenjar kecil yang terletak di pangkal otak — bertanggungjawab mengawal pengeluaran pelbagai hormon. Dalam keadaan normal, ia melepaskan GH mengikut keperluan badan. Pada pesakit akromegali, tumor (adenoma pituitari) tumbuh di dalam kelenjar ini dan mula menghasilkan GH secara berlebihan dan tidak terkawal [5][6].
GH yang berlebihan ini merangsang hati untuk menghasilkan IGF-1 (Insulin-like Growth Factor 1) — hormon yang secara langsung mempengaruhi pertumbuhan tulang, otot, dan organ dalaman. Hasilnya, tulang pada tangan, kaki, dahi, dan rahang terus membesar, manakala organ dalaman seperti jantung turut boleh terjejas. Semua ini berlaku secara sangat perlahan, biasanya bertahun-tahun, menyebabkan ramai pesakit tidak menyedarinya sehingga perubahan sudah ketara.
Punca Akromegali
- Adenoma pituitari — Punca yang paling kerap. Tumor ini bersifat tidak agresif tetapi menghasilkan GH secara berlebihan tanpa kawalan [5][6].
- Tumor ektopik — Dalam kes yang sangat jarang, tumor di luar kelenjar pituitari (contohnya di paru-paru atau pankreas) boleh menghasilkan growth hormone-releasing hormone (GHRH), yang kemudiannya merangsang pituitari untuk menghasilkan terlalu banyak GH.
- Sindrom genetik — Seperti Multiple Endocrine Neoplasia type 1 (MEN1) boleh dikaitkan dengan akromegali, walaupun ini jarang berlaku.
Simptom Akromegali
Kerana perubahan berlaku dengan sangat perlahan, pesakit — malah keluarga mereka — sering tidak menyedari tanda-tanda awal. Kadang-kadang diagnosis baru dibuat apabila seseorang menyedari sarung tangannya tidak muat lagi atau cincin jarinya sudah tidak boleh dipakai.
Antara simptom yang biasa dilaporkan:
- Pembesaran tangan dan kaki — Sering menjadi tanda pertama yang disedari.
- Perubahan wajah — Dahi lebih menonjol, rahang membesar, gigi menjadi renggang, serta bibir atau hidung kelihatan lebih besar.
- Sakit sendi dan badan — Akibat tekanan berlebihan pada tulang rawan dan sendi.
- Kepenatan dan kelemahan otot — Kekuatan otot boleh berkurangan walaupun badan kelihatan lebih besar.
- Sakit kepala — Disebabkan tumor pituitari yang menekan tisu sekitar.
- Gangguan penglihatan — Jika tumor menekan saraf optik, pesakit mungkin kehilangan penglihatan tepi.
- Suara menjadi lebih dalam — Akibat pembesaran sinus dan kotak suara (larynx).
- Sleep apnea — Henti nafas ketika tidur, dikaitkan dengan pembesaran tisu lembut di saluran pernafasan.
- Gangguan metabolisme glukosa — GH yang berlebihan boleh mengganggu fungsi insulin, menyebabkan intoleransi glukosa atau kencing manis [3].
- Peluh berlebihan dan kulit berminyak — Aduan yang kerap tetapi sering diabaikan.
- Tangan atau kaki membesar tanpa sebab yang jelas.
- Perubahan pada rupa wajah yang diperhatikan oleh orang sekeliling.
- Sakit kepala kronik disertai gangguan penglihatan.
- Henti nafas ketika tidur (sleep apnea) yang ketara.
- Paras gula darah tinggi tanpa sejarah kencing manis sebelum ini.
Diagnosis Pembezaan
Tidak semua pembesaran anggota badan atau perubahan wajah menunjukkan akromegali. Doktor perlu mempertimbangkan beberapa keadaan lain:
- Hipotiroidisme — Kekurangan hormon tiroid boleh menyebabkan pembengkakan muka dan kulit kasar yang menyerupai perubahan akromegali.
- Hiperprolaktinaemia — Paras prolaktin yang tinggi boleh hadir serentak dengan akromegali dan kadang-kadang menguasai gambaran klinikalnya, menyebabkan diagnosis akromegali terlewat bertahun-tahun [2].
- Edema akibat penyakit buah pinggang atau jantung — Boleh menyebabkan tangan dan muka kelihatan lebih besar.
- Gigantisme — Jika GH berlebihan berlaku sebelum plat pertumbuhan tulang menutup pada kanak-kanak, hasilnya adalah gigantisme, bukan akromegali.
- Pachydermoperiostosis — Keadaan genetik yang menyebabkan perubahan pada tulang dan kulit menyerupai akromegali tetapi tanpa peningkatan GH.
Bagaimana Akromegali Didiagnosis?
Diagnosis memerlukan gabungan penilaian klinikal dan ujian makmal yang spesifik. Penampilan fizikal sahaja tidak mencukupi untuk mengesahkan diagnosis.
Ujian Darah
- Paras IGF-1 — Ujian tapis utama. Paras IGF-1 yang tinggi mengikut umur dan jantina adalah petunjuk kuat akromegali.
- Ujian penekanan glukosa oral (OGTT dengan GH) — Ujian pengesahan standard. Pada individu normal, minuman glukosa akan menekan paras GH. Pada pesakit akromegali, GH tidak akan turun [6]. Ini adalah ujian paling dipercayai untuk mengesahkan diagnosis.
Pengimejan
- MRI pituitari — Setelah diagnosis biokimia disahkan, MRI dilakukan untuk mengenal pasti saiz dan kedudukan tumor. MRI juga membantu dalam merancang pembedahan dan memantau respons rawatan [1][6].
Pemeriksaan Tambahan
Doktor juga akan menilai komplikasi berkaitan seperti tekanan darah, paras gula darah, fungsi jantung (echocardiogram), dan pemeriksaan medan penglihatan jika tumor besar.
Rawatan Akromegali
Matlamat rawatan adalah untuk mengawal paras GH dan IGF-1 kepada paras normal, mengecilkan atau membuang tumor, dan mengurangkan komplikasi. Pilihan rawatan bergantung kepada saiz tumor, usia pesakit, dan keadaan kesihatan keseluruhan.
1. Pembedahan (Pilihan Utama)
Pembedahan transsfenoidal — iaitu melalui hidung untuk mencapai pituitari — adalah rawatan yang paling disyorkan untuk masalah ini [5]. Kadar kejayaan bergantung kepada saiz tumor; tumor kecil (kurang daripada 10mm) memberikan kadar penyembuhan yang lebih tinggi berbanding tumor yang besar [4]. Pembuangan lengkap sering sukar dicapai apabila tumor sudah besar, dan pendekatan rawatan perlu disesuaikan mengikut keadaan setiap pesakit.
2. Ubat-ubatan
Jika pembedahan tidak berjaya sepenuhnya atau tidak boleh dilakukan, ubat-ubatan digunakan:
- Analog somatostatin (contoh: octreotide, lanreotide) — Meniru hormon semula jadi yang menghalang pengeluaran GH. Berkesan dalam mengawal paras GH dan IGF-1, malah boleh menyebabkan pengecutan tumor dalam sesetengah pesakit [5][6].
- Antagonis reseptor GH (pegvisomant) — Menyekat kesan GH di peringkat reseptor, bukan di pituitari. Data yang ada menunjukkan ia berkesan dalam menormalkan IGF-1 dan memperbaiki toleransi glukosa. Walau bagaimanapun, ia tidak menyekat aktiviti tumor dan kos rawatannya tinggi [3][5].
- Agonis dopamin (contoh: bromocriptine) — Terutama berguna dalam kes tumor campuran yang menghasilkan kedua-dua GH dan prolaktin [6].
- Rawatan kombinasi — Analog somatostatin dan antagonis reseptor GH boleh digunakan bersama untuk kawalan yang lebih baik pada kes yang sukar dikawal dengan satu ubat sahaja [3].
3. Radioterapi
Radioterapi konvensional atau gamma knife radiosurgery dipertimbangkan untuk tumor besar yang tidak boleh dibuang sepenuhnya melalui pembedahan. Kesannya lambat — boleh mengambil masa bertahun-tahun untuk mencapai kawalan hormon — dan hampir semua pesakit akhirnya mengalami kekurangan hormon pituitari (hipopituitarisme) sebagai kesan sampingan jangka panjang [6].
Pemantauan Jangka Panjang
Pesakit akromegali memerlukan pemantauan berterusan walaupun selepas rawatan. Ini termasuk pemeriksaan darah berkala, MRI susulan, dan pemantauan komplikasi seperti kencing manis, tekanan darah, dan masalah jantung. Kawalan yang ketat dilaporkan penting dalam membawa kadar kematian mendekati paras populasi umum [5].
- Berdasarkan saiz dan kedudukan tumor saya, apakah pilihan rawatan yang paling sesuai?
- Adakah saya perlu meneruskan ubat-ubatan selepas pembedahan?
- Berapa kerap saya perlu menjalani ujian darah dan MRI selepas rawatan?
- Apakah komplikasi yang perlu dipantau dalam jangka panjang?
- Adakah ahli keluarga saya perlu menjalani sebarang ujian saringan?
Bukti Secara Ringkas
Kajian-kajian yang tersedia tentang akromegali kebanyakannya adalah kajian retrospektif dengan bilangan pesakit yang kecil — ini tidak menghairankan memandangkan penyakit ini sendiri jarang berlaku. Kadar kejayaan pembedahan yang dilaporkan berbeza-beza mengikut kemahiran pakar bedah, saiz tumor, dan kriteria penyembuhan yang digunakan [4]. Untuk pilihan ubat seperti pegvisomant, data jangka panjang masih terhad walaupun keputusan awal menunjukkan keberkesanan dalam mengawal IGF-1 [3]. Kajian tentang rawatan kombinasi analog somatostatin dan antagonis reseptor GH juga masih terhad kepada kajian kecil [3]. Oleh itu, keputusan rawatan perlu dibuat secara individu bersama pasukan pakar endokrinologi, dengan mengambil kira keadaan klinikal, saiz tumor, dan respons terhadap rawatan.
Rujukan
- Heck A. MR-undersøkelse predikerer behandlingsrespons ved akromegali. Tidsskr Nor Laegeforen. 2016;136(23-24):2035. doi:10.4045/tidsskr.16.0965. PMID: 28004565.
- Dal J, Steffensen C, Hansen TK, Jørgensen JO. Acromegaly masked by symptomatic hyperprolactinaemia. Ugeskr Laeger. 2014;176(1):60-1. PMID: 24629611.
- Jørgensen JO, Feldt-Rasmussen U, Frystyk J, et al. Medical co-treatment of acromegaly with a somatostatin analogue and a growth hormone receptor antagonist. Ugeskr Laeger. 2007;169(10):911-3. PMID: 17359735.
- Madsen M, Matthiesen EB, Poulsen PL, et al. Treatment of acromegaly in Aarhus University Hospital: a retrospective investigation of the period from 1994 to 2004. Ugeskr Laeger. 2007;169(10):907-10. PMID: 17359734.
- Jørgensen JO, Feldt-Rasmussen UF, Andersen M, et al. Acromegaly: new principles for treatment. Ugeskr Laeger. 2007;169(10):904-6. PMID: 17359733.
- Bollerslev J. Acromegaly — diagnosis and treatment. Tidsskr Nor Laegeforen. 2000;120(21):2534-8. PMID: 11070991.